Finding the equilibrium constant of the water gas-shift reaction by direct minimization of Gibbs free energy
The water–gas shift reaction is a gas-phase reaction, widely used in industry, between carbon monoxide and water to form carbon dioxide and hydrogen:
$$ \mathrm{CO(g)} + \mathrm{H_2O(g)} \rightleftharpoons \mathrm{CO_2(g)} + \mathrm{H_2(g)} $$
This equilibrium can be analysed in terms of the Gibbs free energy of its constituents as a function of composition and temperature:
$$ nG_\mathrm{m} = \sum_\mathrm{J} n_\mathrm{J} \left[ RT \Delta_\mathrm{f}G_\mathrm{m, J}^\ominus(T) + RT\ln \left( x_\mathrm{J} \frac{p}{p^\ominus} \right) \right], $$
where \(n_\mathrm{J}\) is the amount of component $\mathrm{J}$, $\Delta_\mathrm{f}G_\mathrm{m, J}^\ominus(T)$ is the standard molar Gibbs free energy of formation of $\mathrm{J}$ at temperature $T$, $x_\mathrm{J} = n_\mathrm{J} / n$ is the mole fraction of $\mathrm{J}$ and $p$ is the total pressure. At the equilibrium composition $nG_\mathrm{m}$ is a minimum.
The formation reactions are:
\begin{align*} \textstyle \mathrm{C(gr)} + \frac{1}{2}\mathrm{O_2(g)} &\rightarrow \mathrm{CO(g)}\\ \textstyle \mathrm{H_2(g)} + \frac{1}{2}\mathrm{O_2(g)} &\rightarrow \mathrm{H_2O(g)}\\ \mathrm{C(gr)} + \mathrm{O_2(g)} &\rightarrow \mathrm{CO_2(g)}\ \end{align*}
so $\Delta_\mathrm{f}G_\mathrm{m, J}^\ominus(T_\mathrm{ref})$ can be calculated for the species $\mathrm{J} = \mathrm{CO(g)}, \mathrm{H_2O(g)}, \mathrm{CO_2(g)}, \mathrm{H_2(g)}$ from tabulated thermodynamic data at the reference temperature $T_\mathrm{ref}=298\;\mathrm{K}$ as $\Delta_\mathrm{f}G_\mathrm{m,J}^\ominus(T_\mathrm{ref}) = \Delta_\mathrm{f}H_\mathrm{m,J}^\ominus(T_\mathrm{ref}) - T_\mathrm{ref}\Delta_\mathrm{f}S_\mathrm{m,J}^\ominus(T_\mathrm{ref})$.
$\Delta_\mathrm{f}G_\mathrm{m,J}^\ominus(T)$ depends on temperature through the Gibbs-Helmholtz equation:
$$ \frac{\mathrm{d}\Delta_\mathrm{f}G_\mathrm{m,J}^\ominus(T) / T}{\mathrm{d}T} = -\frac{\Delta_\mathrm{f}H_\mathrm{m, J}^\ominus(\mathrm{T})}{T^2}. $$
This last equation can be integrated with respect to temperature, with the temperature-dependence of the enthalpy of formation obeying:
$$ \Delta_\mathrm{f}H_\mathrm{m, J}^\ominus(\mathrm{T}) = \Delta_\mathrm{f}H_\mathrm{m, J}^\ominus(\mathrm{T_\mathrm{ref}}) + \int_{T_\mathrm{ref}}^{T} \Delta_\mathrm{f}C_{p, \mathrm{m, J}}^\ominus(T) \,\mathrm{d}T, $$
where $\Delta_\mathrm{f}C_{p, \mathrm{m, J}}^\ominus(T)$ is the difference in heat capacity between species $\mathrm{J}$ and its component atoms in their standard state, e.g. $\Delta_\mathrm{f}C_{p, \mathrm{m, CO}}^\ominus(T) = C_{p, \mathrm{m, CO}}^\ominus(T) - C_{p, \mathrm{m, C}}^\ominus(T) - \frac{1}{2}C_{p, \mathrm{m, O_2}}^\ominus(T)$. The temperature dependence of the heat capacities of individual species is provided by a fit to the the data in the NIST-JANAF Thermochemical Tables:
$$ C_{p, \mathrm{m}}^\ominus(T) = A + Bt + Ct^2 + Dt^3, \quad \mathrm{where} \; t = \frac{T/\mathrm{K}}{1000}. $$
With this in place (and after some calculus), the Gibbs free energy for any composition can be calculated as a function of temperature. The plots of $nG_\mathrm{m}/(RT)$ with composition for four different temperatures are shown below; the equilibrium concentration in each case at is the minimum of these curves.
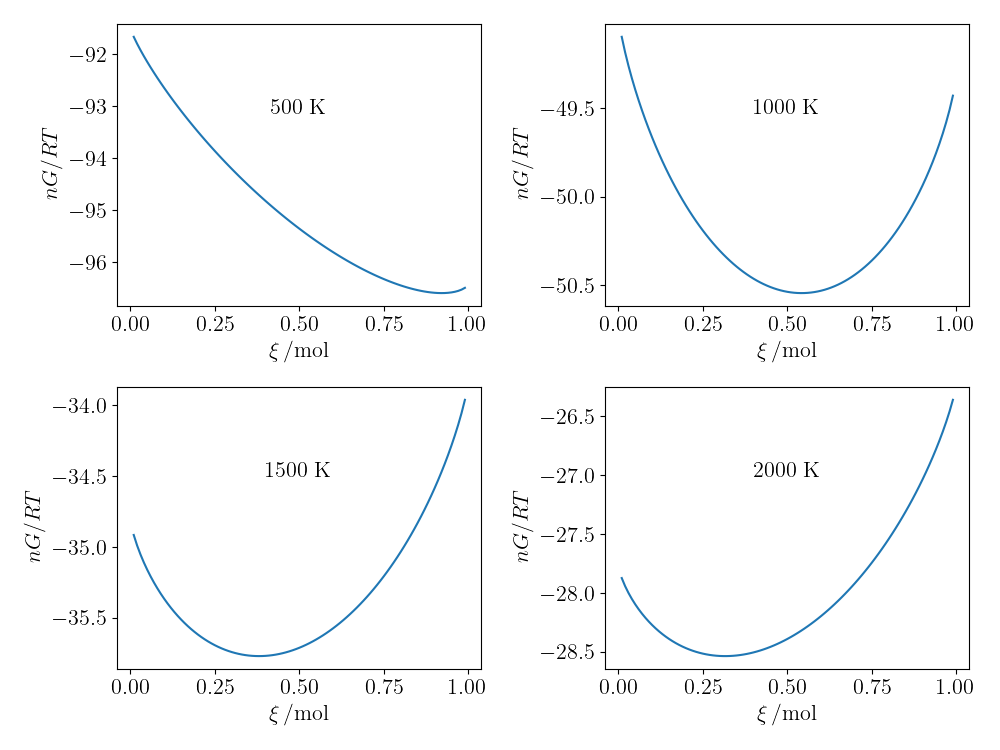
The variation of $nG_\mathrm{m}/(RT)$ as a function of reaction extent (and hence composition) of the water gas-shift reaction for four different temperatures.
This Python code is available on GitHub and as a Jupyter Notebook on MyBinder.
import numpy as np
from scipy.constants import R
import matplotlib.pyplot as plt
Tref = 298.15 # Reference temperature, K
pstd = 1 # Standard pressure, barclass Compound:
"""
A class representing a chemical compound with defined
formula and phase.
"""
def __init__(self, formula, phase, DfHmo, Smo, Cp_coeffs):
"""
Initialize the Compound object with its formula,
phase, standard molar formation enthalpy (kJ.mol-1), standard
entropy (J.K-1.mol-1) and heat capacity temperature-dependence
coefficients.
"""
self.formula, self.phase = formula, phase
self.DfHmo, self.Smo = DfHmo, Smo
self.Cp_coeffs = dict(zip("ABCD", Cp_coeffs))
def __repr__(self):
"""String representation of the Compound."""
return f"{self.formula}({self.phase})"
C = Compound('C', 'gr', 0, 5.6, [8.43, 0, 0, 0])
O2 = Compound('O2', 'g', 0, 205.15, [25.15899701, 15.45182265, -7.08291124, 1.24519819])
H2 = Compound('H2', 'g', 0, 130.68, [29.35864238, -2.30374938, 4.0864225, -0.84549361])
H2O = Compound('H2O', 'g', -241.826, 188.835, [31.65695517, 3.84459406, 8.47064074, -2.77385035])
CO = Compound('CO', 'g', -110.53, 197.66, [27.48525388, 4.25879701, 2.50107857, -1.24204472])
CO2 = Compound('CO2', 'g', -393.52, 213.79, [23.04911248, 56.90377511, -31.87688451, 6.4056037])class Reaction:
"""
A class representing a chemical reaction between
reactant and product Compounds.
"""
def __init__(self, reactants, products):
"""
Initalize the Reaction object with reactants and
products provided as dictionaries of
Compound: stoichiometry key-value pairs.
"""
self.reactants = reactants
self.products = products
# For convenience, create a dictionary of all
# species with positive stoichiometries for products
# and negative stoichiometries for reactants.
self.species = self.products.copy()
for compound, stoich in reactants.items():
self.species[compound] = -stoich
def calc_DrSmo_ref(self):
"""Return the standard molar entropy of reaction in J.K-1.mol-1."""
DrSmo = 0
for compound, stoich in self.species.items():
DrSmo += stoich * compound.Smo
return DrSmo
def calc_DrHmo_ref(self):
"""Return the standard molar enthalpy of reaction in kJ.mol-1."""
DrHmo = 0
for compound, stoich in self.species.items():
DrHmo += stoich * compound.DfHmo
return DrHmo
def calc_DrGmo_ref(self):
"""
Return the standard molar Gibbs free energy in kJ.mol-1 of
reaction at the reference temperature, Tref.
"""
return self.calc_DrHmo_ref() - Tref * self.calc_DrSmo_ref() / 1000# The formation reactants for CO2(g), CO(g), and H2O(g).
CO2_formation = Reaction({C: 1, O2: 1}, {CO2: 1})
CO_formation = Reaction({C: 1, O2: 0.5}, {CO: 1})
H2O_formation = Reaction({H2: 1, O2: 0.5}, {H2O: 1})
formation_reactions = {"CO2(g)": CO2_formation,
"CO(g)": CO_formation,
"H2O(g)": H2O_formation}
# Calculate the standard enthalpies and free energies of
# reaction at temperature Tref. NB these are zero by
# definition for H2(g).
DfHmo_ref = {
"CO(g)": CO_formation.calc_DrHmo_ref(),
"CO2(g)": CO2_formation.calc_DrHmo_ref(),
"H2O(g)": H2O_formation.calc_DrHmo_ref(),
"H2(g)": 0
}
DfGmo_ref = {
"CO(g)": CO_formation.calc_DrGmo_ref(),
"CO2(g)": CO2_formation.calc_DrGmo_ref(),
"H2O(g)": H2O_formation.calc_DrGmo_ref(),
"H2(g)": 0
}def get_DfCp_coeffs(T):
"""
Return the coefficients, valid at temperature T, for the heat capacity
change for the formation reactions of CO(g), CO2(g) and H2O(g) in a
dictionary of species: {"CO(g)": {'A': ..., 'B': ...}, ...}.
"""
# Initialize coefficients to zero.
DfCp = dict((formula, dict((c, 0) for c in "ABCD"))
for formula in ("CO(g)", "CO2(g)", "H2O(g)", "H2(g)"))
for formation_species in ("CO(g)", "CO2(g)", "H2O(g)"):
formation_reaction = formation_reactions[formation_species]
for compound, stoich in formation_reaction.species.items():
for c in "ABCD":
DfCp[formation_species][c] += stoich * compound.Cp_coeffs[c]
return DfCpNow we need the temperature dependence of the Gibbs free energy of formation for each species in the reaction $$ \frac{\Delta_\mathrm{f}G^\ominus_\mathrm{m,J}(T)}{T} = \frac{\Delta_\mathrm{f}G^\ominus_\mathrm{m,J}(T_\mathrm{ref})}{T_\mathrm{ref}} + \Delta_\mathrm{f}H^\ominus_\mathrm{m,J}(T_\mathrm{ref}) \left(\frac{1}{T} - \frac{1}{T_\mathrm{ref}}\right) - g(T) $$ where $$ g(T) = \int_{T_\mathrm{ref}}^T \frac{f(T)}{T^2}\mathrm{d}T, \quad\mathrm{and}\quad f(T) = \int_{T_\mathrm{ref}}^T\Delta_\mathrm{f}C^\ominus_{p,\mathrm{m,J}}(T)\mathrm{d}T. $$
Integrating the Gibbs-Helmholtz equation, $$ \frac{\mathrm{d}\Delta_\mathrm{f}G_\mathrm{m,J}^\ominus(T) / T}{\mathrm{d}T} = -\frac{\Delta_\mathrm{f}H_\mathrm{m, J}^\ominus(\mathrm{T})}{T^2}. $$ from the reference temperature, $T_\mathrm{ref} = 298\;\mathrm{K}$ to the desired temperature gives: \begin{align*} \frac{\Delta_\mathrm{f}G_\mathrm{m,J}^\ominus(T)}{T} - \frac{\Delta_\mathrm{f}G_\mathrm{m,J}^\ominus(T_\mathrm{ref})}{T_\mathrm{ref}} &= -\int_{T_\mathrm{ref}}^T \frac{\Delta_\mathrm{f}H_\mathrm{m,J}^\ominus(T)}{T^2}\mathrm{d}T\\\\ &= -\int_{T_\mathrm{ref}}^T \frac{\Delta_\mathrm{f}H_\mathrm{m,J}^\ominus(T_\mathrm{ref})}{T^2} + \frac{\Delta_\mathrm{f}C_{p, \mathrm{m,J}}^\ominus(T)}{T^2}\mathrm{d}T\\\\ &= \frac{\Delta_\mathrm{f}H_\mathrm{m,J}^\ominus(T_\mathrm{ref})}{T} - \frac{\Delta_\mathrm{f}H_\mathrm{m,J}^\ominus(T_\mathrm{ref})}{T_\mathrm{ref}} - g(T), \end{align*} where $$ g(T) = \int_{T_\mathrm{ref}}^T \frac{f(T)}{T^2}\mathrm{d}T $$ and \begin{align*} f(T) &= \int_{T_\mathrm{ref}}^T\Delta_\mathrm{f}C^\ominus_{p,\mathrm{m,J}}(T)\mathrm{d}T\\\\ & = \Delta A (T - T_\mathrm{ref}) + \frac{1}{2}\Delta B (T^2 - T_\mathrm{ref}^2) + \frac{1}{3}\Delta C (T^3 - T_\mathrm{ref}^3) + \frac{1}{4}\Delta C (T^4 - T_\mathrm{ref}^4). \end{align*} Therefore, \begin{align*} g(T) = &\Delta A \ln \left( \frac{T}{T_\mathrm{ref}}\right) + \frac{\Delta B}{2}(T - T_\mathrm{ref}) + \frac{\Delta C}{6}\left(T^2 - T_\mathrm{ref}^2\right) + \frac{\Delta D}{12}\left(T^3 - T_\mathrm{ref}^3\right)\\\\ & + \left(\Delta A T_\mathrm{ref} + \frac{\Delta B}{2}T_\mathrm{ref}^2 + \frac{\Delta C}{3}T_\mathrm{ref}^3 + \frac{\Delta D}{4}T_\mathrm{ref}^4 \right)\left( \frac{1}{T} - \frac{1}{T_\mathrm{ref}} \right). \end{align*}
def g(T, formation_species, DfCp):
"""
This term in the expression for nG/RT derives from integrating
(twice) the heat capacity change on formation of the component
species with respect to temperature.
"""
DA, DB, DC, DD = (DfCp[formation_species][c] for c in "ABCD")
t, tref = T/1000, Tref/1000
f = 1/t - 1/tref
return (DA * (np.log(t / tref) + tref * f)
+ DB/2 * (t - tref + tref**2 * f)
+ DC/3 * ((t**2 - tref**2) / 2 + tref**3 * f)
+ DD/4 * ((t**3 - tref**3) / 3 + tref**4 * f)
) / 1000
def calc_DfGmo(T, formation_species, DfCp):
"""
Return the standard Gibbs free energy of formation of formation_species
at temperature T from the integrated Gibbs-Helmholtz equation.
"""
DfGmo_over_T = (DfGmo_ref[formation_species] / Tref
+ DfHmo_ref[formation_species] * (1/T - 1/Tref)
- g(T, formation_species, DfCp)
)
return DfGmo_over_T * T# Start with 1 mol of CO(g) and 1 mol of H2O(g) reactants only.
nin = {"CO(g)": 1, "H2O(g)": 1, "CO2(g)": 0, "H2(g)": 0}
def calc_nG_over_RT(xi, T, DfCp):
"""
Return the quantity nG/RT for temperature T and extent of
reaction, xi: xi is the amount of CO(g) reacted.
"""
nout = {"CO(g)": nin["CO(g)"] - xi,
"H2O(g)": nin["H2O(g)"] - xi,
"CO2(g)": nin["CO2(g)"] + xi,
"H2(g)": nin["H2(g)"] + xi}
ntot = sum(nout.values())
nG_over_RT = 0
for species in ("CO(g)", "H2O(g)", "CO2(g)", "H2(g)"):
DfGmo = calc_DfGmo(T, species, DfCp)
x = nout[species] / ntot
nG_over_RT += nout[species] * (DfGmo / R / T * 1000 + np.log(x * pstd))
return nG_over_RTdef plot_nG_over_RT(T, ax):
"""Plot nG/RT vs. xi on Matplotlib Axes ax."""
DfCp = get_DfCp_coeffs(T)
xi_grid = np.arange(0.01, 1, 0.01)
nG_over_RT = np.zeros(xi_grid.shape)
for i, xi in enumerate(xi_grid):
nG_over_RT[i] = calc_nG_over_RT(xi, T, DfCp)
ax.plot(xi_grid, nG_over_RT)
ax.set_xlabel("xi")
ax.set_ylabel("nG/RT")
ax.text(0.5, 0.7, f"{T} K", transform=ax.transAxes, ha="center", va="center")fig, axes = plt.subplots(nrows=2, ncols=2)
plot_nG_over_RT(500, axes[0][0])
plot_nG_over_RT(1000, axes[0][1])
plot_nG_over_RT(1500, axes[1][0])
plot_nG_over_RT(2000, axes[1][1])
plt.tight_layout()The equilibrium position of the composition occurs at the minimum of $nG_\mathrm{m}$, which can be found numerically using scipy.optimize.minimize, constraining the reaction extent (number of moles of CO reacted) to be between 0 and 1 mol, since there is 1 mol initially.
from scipy.optimize import minimize
def get_K(T):
DfCp = get_DfCp_coeffs(T)
cons = ({'type': 'ineq', 'fun': lambda X: X[0]},
{'type': 'ineq', 'fun': lambda X: 1-X[0]})
res = minimize(calc_nG_over_RT, (0.1,), args=(T, DfCp), method="slsqp",
constraints=cons)
x = res.x[0]
K = x**2 / (1-x)**2
return KK = []
for T in (500, 1000, 1500, 2000):
K.append(get_K(T))
K[137.03092238027537,
1.4040566158667849,
0.37749729566513246,
0.2128298922078678]The comparison with the following fitted function given by Catilin Callaghan in her 2006 PhD thesis is pretty good:
$$ \log_{10} K = -2.4198 + 0.0003856T + \frac{2180.6}{T}. $$
def Keq(T):
return 10**(-2.4198 + 0.0003856*T + 2180.6/T)
for T in (500, 1000, 1500, 2000):
print(Keq(T))136.20717952990907
1.4008769839545847
0.40957489947329023
0.276503096983568